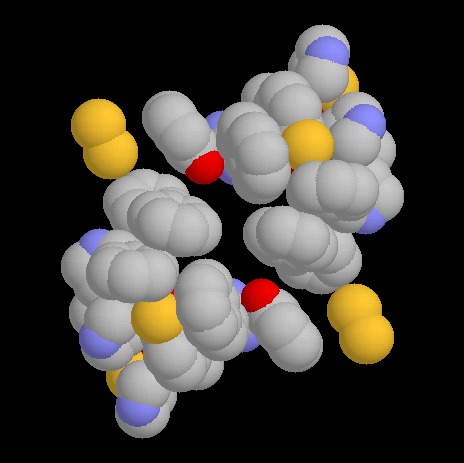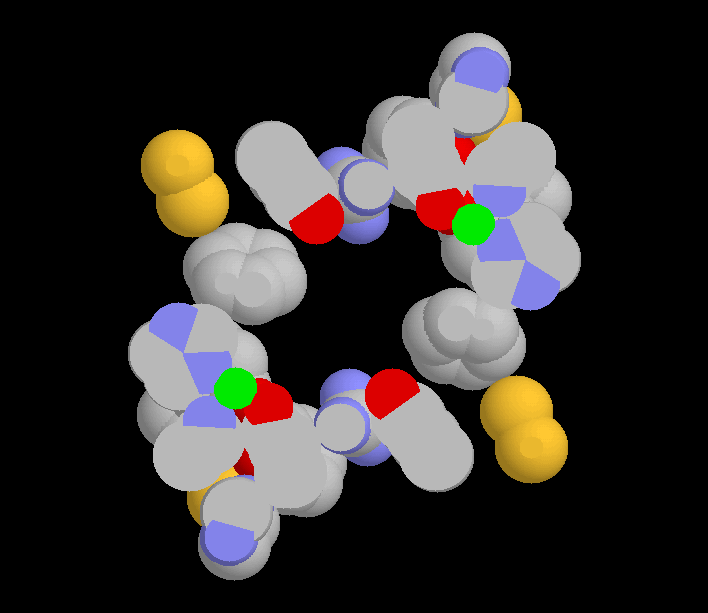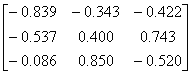
Proposed tetrahedral quaternary structure for Galactose Oxidase.
by Donald G Vanselow
Introduction
Galactose oxidase is an enzyme
produced by some filamentous fungi. It has found application in
biochemical research (1, 2) and potential uses in food technology
(3). It is thought that its physiological role is to assist in
degradation of the complex solid energy substrates of these fungi
(4). The enzyme occurs in intracellular and extracellular forms (5).
Galactose oxidase is reported to be a monomeric enzyme of molecular weight 68000 with one copper atom per protein molecule. The crystal structure of the extracellular form has been reported (6) and its largest domain resembles the propeller structure of neuraminidase, but with seven "blades" instead of six.
There are a number of anomalous features of the enzyme that are difficult to reconcile with the reported monomeric state.
Enzyme preparations harvested from different batches of fungal growth differ widely in the proportion of enzyme that is catalytically active (7). It is not clear why the fungus would produce inactive enzyme, unless the inactive form was smaller than the active form and therefore could diffuse more readily through complex substrates.
The enzyme is reported to be robust, remaining active in 6M urea (8). On the other hand, it easily loses its activity during storage (9), and dilution and purification (8). It also gains activity on exposure to peroxidase (9) or certain oxidants (7). This kind of behaviour suggests dissociation but not denaturation of subunits.
The intracellular form displays cooperative binding of either galactose or oxygen (5), but is thought to have only one binding site. It is generally accepted that cooperative binding requires the cooperating sites to be in the same molecular assembly.
The enzyme catalyses a two-electron reduction of O2 to H2O2 and two-electron oxidation of a substrate alcohol to an aldehyde, but the only cofactor is a single copper atom that can only donate or accept a single electron (7).
Much research has been directed towards exploration of the mechanism of this enzyme because it appears to be structurally simpler than other enzymes that catalyse reaction with molecular oxygen (10). However, the kinetic behaviour of enzyme preparations is even more complex than foreshadowed above, showing lags and surges and non-linear dependence on enzyme concentration (10).
Hamilton et al. (10) considered the possibility that the active form of the enzyme was a dimer or higher polymer and compared the ultracentrifuge sedimentation rates of activated and non-activated enzyme. The rates were almost the same and the authors concluded that there was no evidence of polymerisation. They ignored the possibility that the polymer, if any, might have differed in shape from the monomer and consequently could have had a sedimentation rate not very different from the monomer. (Without shape effects, the increased effective weight of a polymer normally overcomes the increased frictional drag on the polymer and it sediments at a faster rate.) No other authors have considered the possibility that the protein of MW 68000 is a subunit of a larger catalytically active structure. The main reason for this is that there has been only one report of an electrophoretically mobile band with MW of about 300000 (8), which would correspond to a tetramer. On the other hand, a number of other methods of estimating molecular weight in solution have produced results consistent with a value near 68000 (8). However, related studies (8, 10) also showed that most of the enzyme in those preparations was not catalytically active but could be brought into an active state by the presence of peroxidase and non-saturating levels of galactose or by oxidation by ferricyanide. In the work of Hamilton et al. (10), one enzyme preparation was only about 3% in the active form. This leaves open the possibility that the active form of galactose oxidase is a tetramer that was not detected by physical methods. It may be that it is advantageous to the fungus to produce a tetramer that readily dissociates into monomers. These would more easily penetrate the complex solid substrates on which the fungus grows.
Mendonca and Zancan (5) measured the elution volume of intracellular galactose oxidase in a gel filtration experiment. They assayed the enzyme by reaction with peroxidase and galactose and compared the elution volume with elution volumes of other proteins of known molecular weights. They thus estimated the molecular weight of galactose oxidase to be near 65000. In quoting this result I am assuming that the intracellular form of the enzyme does not greatly differ from the extracellular form that is the subject of the present discussion. Like the ultracentrifugation experiment of Hamilton et al. (10), this came close to simultaneous determination of molecular weight and enzyme activity. However, it could be that the assay conditions, involving peroxidase and galactose, brought about assembly of the catalytic form from inactive monomers eluted from the column. Again the question of whether the active form of galactose oxidase is a tetramer remains open.
The aim of the present work was to arrange the crystal structure of galactose oxidase into a plausible tetrahedral form and relate the new structure to the enzyme’s chemical and biochemical properties.
Method
The crystal structure of galactose oxidase (6) was obtained from
the Protein Data Base (record number 1GOF). The propeller domain was
manually fitted into a tetrahedral arrangement in the manner
previously described for neuraminidases (11). The two additional or
auxilliary domains were found not to clash with the tetrahedral
structure and were allowed to remain in the same relative positions
found in the crystal.
Results
and Discussion
The
tetrahedral structure can be obtained by first rotating 1GOF with the
matrix
and then adding 44.354, 8.672 and
22.724 Ångstrom to the x, y and z coordinates
respectively
This subunit was designated Chain A. Three
symmetry-related copies of Chain A were then generated by rotating it
180° about the x, y and z axes respectively. These three subunits
were designated Chain B, Chain C and Chain D respectively.
As for the neuraminidases (11), the subunits were orientated so that the active site of Chain A was occluded by Chain D and vice versa. The gradient of the propeller axis of Chain A in the xy plane is less than one and the proximal end of the axis is above the xz plane, as for the neuraminidases. It is equally possible that an arrangement with the axis gradient more than one and the proximal end below the xz plane could produce a suitable interface between Chains A and D. Such a structure would be a pseudo mirror-image of the neuraminidases. This does not occur in this case. As yet it cannot be said whether the observed similarity to the neuraminidases is coincidence or not.
Figures 1, 2 and 3 are views of the assembled tetramer seen along the coordinate axes.
|
|
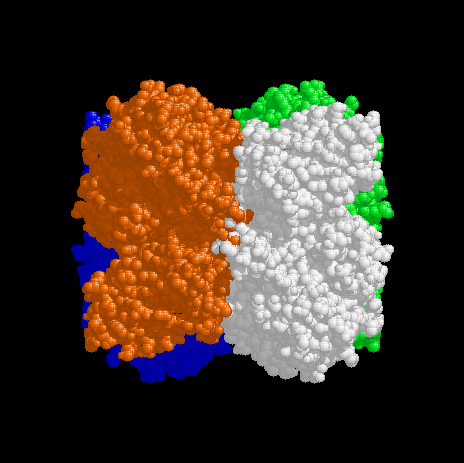
Figure 1. View of the tetramer down the x axis. The y axis is up and the z axis to the left. Chain A is red, Chain B white, Chain C green and Chain D blue. The parts of the A (red) and B (white) subunits at the bottom left and top right of the tetramer are the N-terminal domains. A small amount of steric clashing can be seen in the centre of the figure involving residues 148-153. Perhaps these residues have been moved during crystallisation and belong in the adjacent shadowed cavities. |
|
|
|
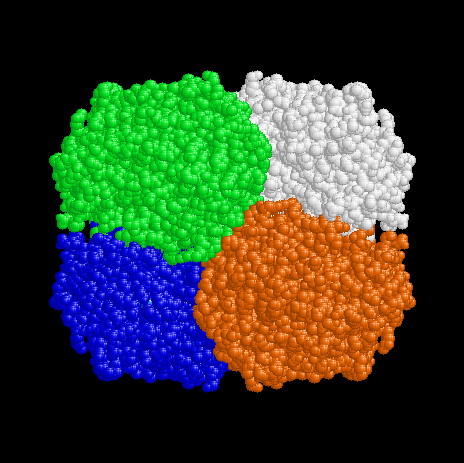
Figure 2. View of the tetramer down the y axis. The x axis is to the right and the z axis is down. Chain A is red, Chain B white, Chain C green and Chain D blue, as for Fig 1. |
|
|
|
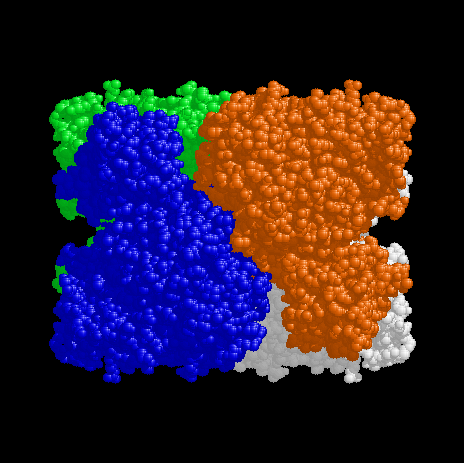
Figure 3. View of the tetramer down the z axis. The x axis is to the right and the y axis is up. Chain A is red, Chain B white, Chain C green and Chain D blue, as for Fig 1. The parts of the D (blue) and A (red) subunits at the top left and bottom right of the tetramer are the N-terminal domains. The metal centres are buried near the interface between Chains A and D (or Chains B and C). This view corresponds with the view of Figures 4 and 5 below. |
It is interesting to note that this tetrahedral arrangement of subunits brings the region of delocalised π-electrons in each subunit into contact with the same region of a neighbouring subunit. Figure 4 shows the aromatic rings surrounding the copper atoms of Chain A and Chain D. The aromatic rings are from Phe194, Phe227, Tyr272, Trp290, Tyr329, His334, Tyr405, Phe441, Phe464, Tyr495, His496 and His581. Also included are the sulfur atoms of Cys228, Cys383, Cys515 and Cys518 as well as the guanidino group of Arg330. All these groups have π-electron or non-bonding electron pair clouds in contact with each other, with atom centres 4 Å or less apart. They are here considered to be a single aromatic system held together by constraint (12) and coordinated to the copper atom.
Figure 5 is the same view as shown in Figure 4 but as a slab section. This allows the copper atoms and the active sites to be seen.
|
|
Figure 4. The aromatic systems surrounding the copper atoms of Chains A and D, viewed down the z axis. The y axis is up and the x axis to the right, as in Fig 3. Carbon is grey, oxygen red, nitrogen blue and sulfur yellow. The two aromatic systems come close enough together to form one greater system. |
|
|
|
Figure 5. A slab section through the aromatic systems of Chains A and D so that the copper atoms (green) can be seen. The view is down the z axis as for Fig 4. The vacant space between the copper atoms is the proposed active site (4). The space connects to the exterior of the enzyme either along the z axis, where only side-chains block access, or via channels along the pseudo-threefold axes where three subunits meet. These channels are also partly blocked by sidechains. |
In Figure 5 the active site in the centre must connect by a channel to the exterior of the tetramer. As for the neuraminidases (11), such a channel is required if the enzyme is to act on a substrate structure while it is part of a polysaccharide or glycoprotein. Indeed, galactose oxidase does oxidise the terminal galactose of such polymers (1, 4, 9). There seem to be two possible sites of channels but both are partly blocked by sidechains.
In Figure 4 the closest approach between the aromatic systems of chains A and D is a separation of about 1 Å between Cys515 of one chain and Tyr329 of the other (4.8 Å between atom centres). This small separation is an artefact of the intrinsic softness of the protein and the incomplete docking achieved by the manual method employed here. It seems that for part of the reaction cycle of this enzyme, copper atoms of adjacent subunits are coordinated into a greater aromatic system enabling the relay of electrons between the two metal centres. Such an arrangement could facilitate two-electron oxidations and reductions without the necessity of postulating the conversion of an amino acid into a free radical, as proposed by Ito et al. (6). In addition, two disulfide bonds (Cys515 to Cys518) contact the π-electron cloud allowing scope for wider coupling of redox changes under some conditions.
To maximise the rate of two-electron reduction and oxidation, efficient coupling between the two copper atoms would be required for a substantial fraction of the time. That is, it would seem to be important that electrons could pass to both copper atoms simultaneously, and, in the face of thermal motion, it is suggested that the tetrahedral structure provides the required rigidity to keep the copper atoms in electronic contact for a considerable fraction of time. The relationship between tetrahedral structure and rigidity is described in the theory of constraint (12).
Recognition of the role of the whole aromatic system in catalytic function might also allow understanding of the results of protein chemical studies of the active site (4).
Conclusion
The tetrahedral structure is a good fit that is compatible with the locations of the auxilliary domains. It provides an explanation for all the anomalous features of the enzyme, outlined in the Introduction, and could provide a rigid core to support the catalysis of reduction and oxidation according to the theory of constraint (12).
References
(1) Avigad, G. (1985). Oxidation rates of some desialylated glycoproteins by galactose oxidase. Arch. Biochem. Biophys., 239, 531-537.
(2) Kelleher, F. M., Dubbs, S. B. and Bhavanandan, V. P. (1988) Purification of galactose oxidase from Dactylium dendroides by affinity chromatography on melibiose-polyacrylamide. Arch. Biochem. Biophys., 263, 349-354.
(3) Wilkinson, D., Akumanyi, N., Hurtado-Guerrero, R., Dawkes, H., Knowles, P. F., Phillips, S. E. V. and McPherson, M. J. (2004) Structural and kinetic studies of a series of mutants of galactose oxidase identified by directed evolution. Protein Eng. Des. Selection , 17, 141-148.
(4) Knowles, P. F. and Ito, N. (1993) Galactose Oxidase. In Perspectives on Bioinorganic Chemistry. Hay, R. W., Dilworth J. R. and Nolan, K. B. Eds.; JAI Press: London, pp 207-244.
(5) Mendonca, M. H. and Zancan, G. T. (1987) Purification and characterization of intracellular galactose oxidase from Dactylium dendroides. Arch. Biochem. Biophys., 252, 507-514.
(6) Ito, N., Phillips, S. E. V., Stevens, C., Ogel, Z. B., McPherson, M. J., Keen, J. N., Yadav, K. D. S. and Knowles, P. F. (1991) Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature, 350, 87-90.
(7) Whittaker, M. M. and Whittaker, J. W. (1988) The active site of galactose oxidase. J. Biol. Chem., 263, 6074-6080.
(8) Kosman, D. J., Ettinger, M. J., Weiner, R. E. and Massaro, E. J. (1974) The molecular properties of the copper enzyme galactose oxidase. Arch. Biochem. Biophys., 165, 456-467.
(9) Tressel, P. S. and Kosman, D. J. (1982) Galactose oxidase from Dactylium dendroides. Method Enz., 89, 163-171.
(10) Hamilton, G. A., Adolf, P. K., de Jersey, J., DuBois, G. C., Dyrkacz, G. R. and Libby, R. D. (1978) Trivalent copper, superoxide, and galactose oxidase. J. Am. Chem. Soc., 100, 1899-1912.
(11) Vanselow, D. G. (2007) Tetrahedral structures in the neuraminidases with special reference to influenza neuraminidase. Available to download from http://nativeproteins.blogspot.com
(12) Vanselow, D. G. (2002) Role of constraint in catalysis and high-affinity binding by proteins. Biophys. J. 82, 2293-2303. Internet access via http://nativeproteins.blogspot.com
Please
send me your comments, suggestions or questions.
mailto:dvanselow@hotmail.com