Tetrahedral
structures in the neuraminidases with special reference to influenza
neuraminidase.
Donald G.
Vanselow
54 Greenways Road,
Glen Waverley, Victoria 3150,
Australia
Keywords
Structure-function,
Solution structure, Crystal structure,
Symmetry, Constraint, Natural selection
Running title
Tetrahedral
structures in neuraminidases
Other address for correspondence
email: d.vanselow@UNSWalumni.com
Return
to http://nativeproteins.blogspot.com
ABSTRACT This report
shows that a rearrangement of subunits from enzyme crystal
structures, guided by symmetry and biophysical requirements, leads to
a set of new tetrahedral quaternary structures for neuraminidases
from viruses, bacteria and higher organisms. The new structures
provide constraining environments for the active sites. In the case
of viral neuraminidases, the locations of glycosylation sites and the
points of attachment of the stalk or tether, together with the
quality of steric and electrostatic fit, provide strong evidence that
the tetrahedral structure is a native structure. For those
neuraminidases from bacteria and the leech that have additional
domains, the steric fit with the extra domains is also consistent
with these being native structures. This discovery confirms the role
of constraint in enzyme function and suggests the basic features to
be expected in native structures of other enzymes and proteins.
Introduction
In an earlier study of the theory of
protein structure and function (1) it was argued that an enzyme must
be able to constrain its active site. Using approximate calculations
it was shown that the size of enzymes and the relative abundance of
tetrameric forms were consistent with the provision of
three-dimensional constraint. The theory predicts that the catalytic
form of a tetrameric enzyme will be a tetrahedral arrangement of
subunits with centrally located active sites. However, it was noted
that the structures of proteins in crystals, on first inspection,
showed no evidence of such arrangements.
In this
study the aim was to explore alternative arrangements of subunits of
a well-characterized family of enzymes composed of only one type of
subunit. Some of the enzymes were reported to be homotetrameric, and
some had been reported to have other degrees of association, based on
associations seen in crystals. However, given that the assumption
underlying the aim was that associations observed in crystals may not
be the only or the principal native quaternary structures, all the
enzymes were subjected to the same search for tetrameric forms. In
general, a simultaneous four-molecule docking problem has a
prohibitively large number of degrees of freedom and cannot be
solved. However, consideration of the principles of evolution and
Natural Selection as detailed in Appendix 1 suggests that a
tetrahedral homotetramer should have D2 symmetry in its native state
and the degrees of freedom become manageable. That is, by choosing a
family of enzymes with only one type of subunit, the number of
possible arrangements to be explored is greatly reduced. In addition,
the viral enzymes have surface glycosylation sites in many cases, as
well as a stalk or tether attachment, the locations of which further
limit the possible quaternary structures. The neuraminidase
superfamily (2) occurs across a diverse range of organisms. All
members have in common a six-bladed “propeller” domain
containing the active site, but may or may not have other loops or
domains attached. The present study shows that, across the whole
superfamily, there is a common tetrahedral arrangement of the
propeller domains consistent with the locations of viral tethers and
glycosylations as well as the locations of various attached auxiliary
domains and loops.
The
best-known neuraminidases, those from the influenza virus, are then
studied in detail. Various experimental observations from the
literature are explained in terms of the tetrahedral structure.
Methods
Docking.
For each neuraminidase, the docking was
primarily focused on the propeller domain. Sugar moieties or
auxiliary domains attached to the propeller domain by a single
freely-rotatable bond were considered to be mobile and their
orientations in the crystals were discounted. In practice these
appendages were deleted from the crystal structure files before
docking was attempted. After docking, their points of attachment were
checked for access to the solvent space. Loops or domains attached by
two single bonds were considered to have a fixed orientation relative
to the propeller and were not deleted before docking. Side chains of
surface amino acids were considered to have a large degree of freedom
of orientation and apparent clashes between them were given reduced
weight in line with the practice of “soft docking” (3).
Docking was
achieved by a computer-assisted manual trial-and-error method. To
save time it was essential to use as much prior information as
possible to narrow the search. Consequently, the docking was
attempted first on the viral enzymes because of the relatively large
number of surface features (tether and sugar side-chains) providing
clues to the native orientation. After it became apparent that
certain features of the propeller orientation were conserved, this
orientation was used as the starting point for docking the bacterial
and animal neuraminidases. Initially the quality of fit in the
tetramers was judged subjectively by the lack of empty space between
subunits and the lack of steric clashes, especially backbone clashes.
Subsequently methods were developed to quantify these features. In
all, 11 neuraminidases were reassembled into tetrahedral structures.
The first
neuraminidase reassembled was the influenza virus N2 type. The atomic
coordinates of the crystal structure of this enzyme, are recorded in
the Protein Data Base (PDB) with the identification code 1NN2 (4).
Using the Swiss-PdbViewer (version 3.7) (5), the propeller axis (4)
of a subunit of N2 neuraminidase was aligned along the (1, 1, 1) axis
with the active site facing towards the origin. The (1, 1, 1) axis
was chosen for convenience so that the two-fold rotational symmetry
axes of the tetramer would be the x, y and z axes. The subunit was
rotated about the (1, 1, 1) axis until Cysteine92 was as close as
possible to the xy plane and the new coordinates were saved as a PDB
file. The disulfide bond between Cys92 and Cys417 is the point of
covalent attachment of the stalk or tether to the catalytic domain of
the enzyme, as discussed in Appendix 2. For reasons discussed in
Appendix 2, the most probable location of the point of attachment in
the native structure would be on a plane containing two symmetry
axes, and this was chosen to be the xy plane. The tether and the
oligosaccharides attached to certain asparagine residues were then
deleted from the PDB file.
Three more copies of the
relocated and edited N2 structure were then rotated 180°
about the x, y and z axes respectively to generate
a tetramer with D2 symmetry. The tetramer was then examined visually
for quality of steric fit. The process was repeated several times
with minor adjustments to the alignment of the propeller axis, the
rotation about the (1, 1, 1) axis and the distance from the origin,
to simultaneously optimize the three interfaces of each monomer with
its neighbours. For convenience in describing relationships between
subunits in the tetramer, the subunit aligned along the (1, 1, 1)
axis was designated the A chain, while the copies rotated about the
x, y and z axes were designated chains B, C and D respectively.
Similar procedures and conventions were used for the docking of all
11 proteins.
Recording of atomic
coordinates The
atomic coordinates of the repositioned subunits are recorded in
Appendix 3 as the translational and rotational operations that need
to be applied to the respective original coordinates from the Protein
Data Base to generate chain A of the tetrahedron.
Description of propeller
orientation. For each protein, the axis
of the propeller was defined as a line passing through the
catalytically active tyrosine oxygen atom in the active site (6, 7),
at the proximal end, and through the mid-point of the propeller
blades at the distal end. This mid-point was the average of the
coordinates of the alpha carbons of the six amino acids, one from
each blade, nearest the axis at the distal end. The relevant amino
acids are listed in Table 1. In addition to defining the propeller
axis it was necessary to define the rotational orientation of the
propeller about its axis. The catalytically active tyrosine described
above has its phenolic oxygen on the propeller axis and its alpha
carbon embedded in the sixth blade of the propeller in all the
neuraminidases studied. Thus, the axis through the phenolic oxygen
and the alpha carbon, the tyrosine axis, characterizes the rotational
orientation of the propeller. The orientation of the tyrosine axis
relative to the y axis was calculated by projecting both those axes
onto a plane normal to the propeller axis and measuring the clockwise
angle from the projected y axis to the projected tyrosine axis as
viewed towards the origin. In practice this was done by translating
the propeller so that its axis passed through the origin, then
rotating it about y and then about z so that the propeller axis lay
along the x axis. Thus the y axis became normal to the propeller axis
and the slope of the tyrosine axis was calculated from the y and z
coordinates of its alpha carbon.
Measurement of effect of
propeller rotation.
The docking results showed that the
main differences in propeller orientation between species were in the
rotational orientation about the propeller axis. Therefore, for three
neuraminidases, other rotational orientations about the propeller
axis were searched to confirm that the observed orientation of best
fit was indeed unique with respect to that rotational degree of
freedom. Angle of rotation was measured in the same way that
propeller orientation, above, was measured. Lack of fit was measured
at two levels of sensitivity; the number of atoms of any type
apparently clashing, and the number of backbone atoms apparently
clashing with other backbone atoms.
Detection of apparent steric
clashes
The in-built algorithm of RASMOL (8)
was used for measuring the notional clashes generated by rotation of
subunits about the propeller axis, with 3.0 Å separation
between atomic centers taken as the threshold for a clash. In the
detailed study of N2 and N9 neuraminidases, apparent steric clashes
between subunits were detected using the CONTACT program of the CCP4
suite (9).
Improved docking of N2 and N9
neuraminidases To
illustrate the ease with which the apparent clashes between side
chains could be avoided in the proposed neuraminidase structures,
selected rotamers from the Swiss PDBViewer library were substituted
into the structure and found to largely eliminate the clashes. The
substitutions in N2 were (rotamer library numbers in square
brackets), Asp147 [6], His150 [8], Arg152 [18], Trp178 [9], Asp198
[3], Leu321 [1], Asp330 [2], Arg331 [12], Asn342 [15], Lys431 [7],
and Thr455 [2]. In N9 the substitutions were Asn146 [2], His150 [7],
Arg152 [18], Asn345 [10], Trp437 [3] and Gln455 [8]. Their
coordinates in the A chain are included in the Supplementary
Material in PDB format. The substitutions are not used elsewhere
in this paper, except in the determination of buried surface areas
detailed below.
Measurement of accessible and
buried surface areas The
AREAIMOL program of the CCP4 suite (9) was used to calculate the
accessible surface areas of monomers and tetramers of N2 and N9
enzymes, with various probe sizes. From these measurements, the
buried surface areas were calculated.
Results and Discussion
Tetrahedral
structures
Appendix 3 lists the transformations
required to generate the A chain of each tetrahedron from the
published coordinates in the Protein Data Base. The B, C and D chains
can be generated by rotating the A chain 180°
about the x, y and z axes respectively.
For N2 and N9, the only apparent steric clashes between main-chain
atoms are between pairs of Asn342 residues on adjacent N2 chains and
pairs of Gly343 on N9 chains. These apparent clashes could be
relieved by displacements of about 1 Å. The position of Asn342
in the X-ray crystal structure of N2 is among the most uncertain (4),
and Gly343 lies in a region of high mobility in the N9 crystal
structure (10). The clashes are described as "apparent"
because, if the subunits were brought together as described, the
clashes would be avoided by very low energy conformational changes.
Such low energy conformation changes have been observed in the
surface residues of proteins crystallized with and without another
protein, as discussed by Del Carpio Munoz et al. (3). Provision for
the conformation changes is now incorporated in the techniques of
"soft docking" (3, 11, 12). Del Carpio Munoz et al. (3)
have allowed for the softness of protein surfaces by treating the
outer 2.2 Å of protein surfaces as plastic materials able to
freely interpenetrate each other.
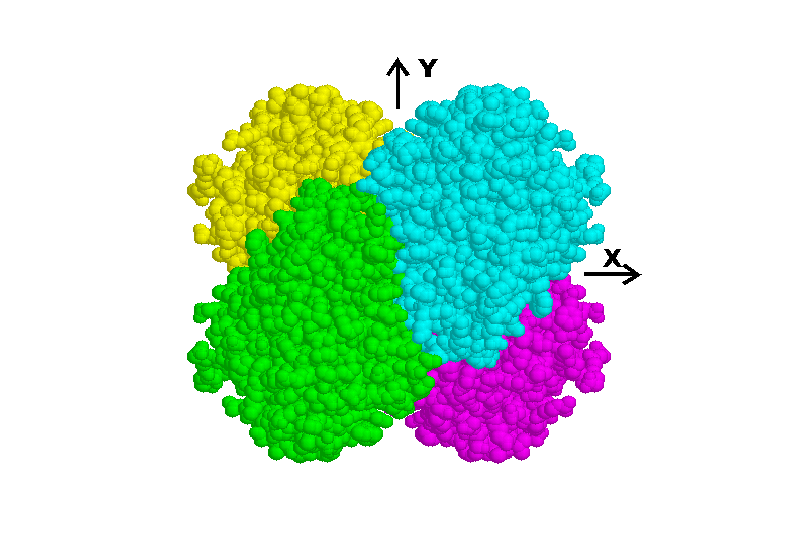
Figure 1 is a computer graphic of the
proposed N9 tetramer viewed down the y axis, showing the proximity of
Cys92 residues to the xy plane. Figure 2 is a view down the z axis
showing the self-complementarity of the interfaces.
|

|
Fig 1. Tetramer of N9-type subunits, viewed down the y axis.
Cys92 residues, coloured black and marked by arrows, lie close to
the xy plane, indicated by the horizontal line. The figure was
produced using RASMOL (8)
|
|
|
|
Fig 2. Tetramer of N9-type subunits viewed down the z axis. The
self-complementarity of the two foreground subunits can be seen in
the curved interface near the center of the figure. The figure was
produced using RASMOL (8).
|
|
The
Influenza B enzyme crystal structure does not dock with itself quite
as well as N2 and N9 do in the region of the active site, but it is
otherwise similar to the other two influenza enzymes. There are no
clashes between backbone atoms.
Parainfluenza-3
neuraminidase docks with itself very well and similarly shows no
clashes between backbone atoms.
The
parainfluenza-5 neuraminidase crystal structure lacks coordinates for
residues 187-190. These amino acids would lie at the interface near
the active site and therefore that part of the docking cannot be
assessed. Apart from the missing residues, the docking is very good.
The crystal
structure of the neuraminidase of Newcastle Disease Virus does not
dock very well compared with the other viral neuraminidases. The
tabulated transformations represent the best docking achievable
without moving certain loops, especially residues 518-521. This loop
has substantially different conformations in other PDB files of the
same protein, but none of the files produces better docking.
Among the
bacterial neuraminidases, Salmonella neuraminidase has only the
propeller domain and it docks with itself fairly well, although some
movement of residues 332-334 would improve the docking.
The Vibrio
neuraminidase has two auxiliary domains in addition to the propeller
domain. The N-terminal domain, joined to the propeller at residue
214, would clash in the tetrahedral structure but may be rotated out
of the way. The second auxiliary domain is a loop from residues 354
to 539. It does not clash in the tetrahedral structure and
contributes to the complementarity of docking.
Micromonospora
viridifaciens has a neuraminidase with two C-terminal auxiliary
domains. The first is joined to the propeller by residue 403 as well
as a disulfide link from 351 to 405. The second is joined to the
first through residue 503. This neuraminidase provides the most
compelling visual evidence that the tetrahedral structures are
natural, because of the complex shape of the monomer and the
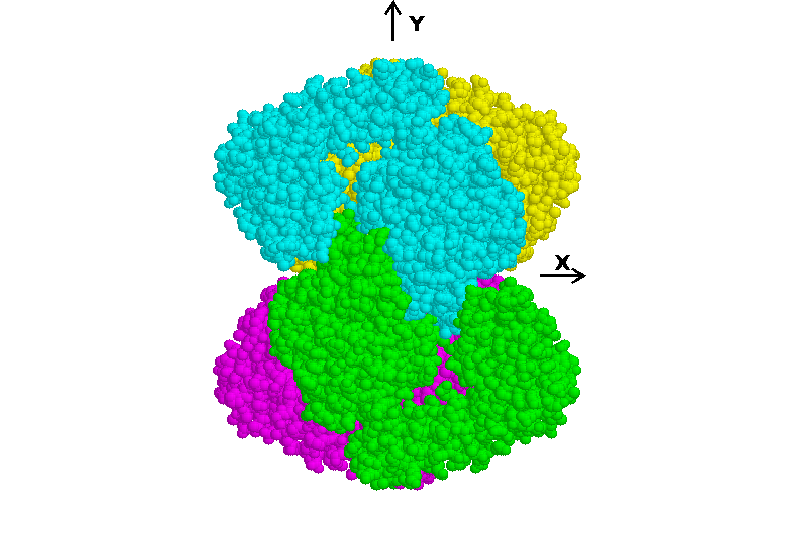
interlocking nature of the tetrahedral structure. Figure 3 is a view
of the proposed tetramer viewed down the z axis.
|
|
Fig 3. Tetramer of Micromonospora subunits viewed down the z
axis. Two propeller domains can be seen docked with each other in
the center. The attached C-terminal domains wrap around to dock
with the symmetry-related propeller domain. The figure was
produced using RASMOL (8).
|
|
Comparison with Figure 2 shows that the
docking of the propeller domains of chain A and chain D is the same
in influenza and Micromonospora, despite the 86°
difference in rotational orientation of the
propellers (Table 1). This docking of the four central propeller
domains of Micromonospora coincides with remarkably good docking of
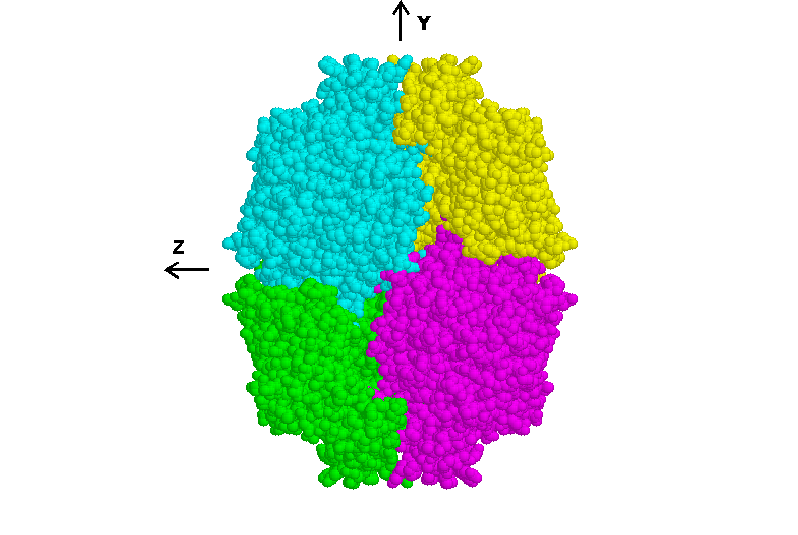
the other eight domains. The quality of the fit is further
illustrated in Figure 4, which is a view down the x axis of the
Micromonospora tetramer.
|
|
Fig 4. Tetramer of Micromonospora subunits viewed down the x
axis. This view again shows the high quality of docking. The
figure was produced using RASMOL (8).
|
|
For Micromonospora, the docking was initially carried
out on the propeller domains alone and it was only subsequently
discovered that the same transformation produced very good docking of
all domains.
The Leech
neuraminidase has an N-terminal auxiliary domain joined to the
propeller through Glycine277. This domain extensively overlaps with
the propeller domain of symmetry-related subunits across the x axis,
but can be rotated out of the way. There is also an auxiliary loop
domain from residue 406 to 504 that docks well with its
symmetry-related counterpart across the z axis. These domains
contribute to the occlusion of the active site.
The Human
neuraminidase has no auxiliary domains and docks well across the x
and y axes. However a large number of residues near the z axis could
not be located by X-ray crystallography and so the docking near the
active site cannot be assessed.
Geometry of the tetrahedral
structures
Table 1 summarises the geometrical characteristics of the
neuraminidase tetramers.
TABLE 1
Geometrical parameters in the tetrahedral structures.
|
Enzyme type
|
Propeller axis
|
Tyrosine axis
|
|
|
Distal end
|
Proximal end
|
Equations to axis
|
|
|
|
Residue Nos.
|
Tyr No.
|
y=mx+c
|
z=nx+d
|
*Angle from y
|
|
|
|
|
m
|
c (Å)
|
n
|
d (Å)
|
|
|
Influenza N2
|
126, 184, 233,
284, 356, 413
|
406
|
0.64
|
5.1
|
0.01
|
17.0
|
152°
|
|
Influenza N9
|
126, 185, 233,
283, 356, 412
|
406
|
0.62
|
4.9
|
0.05
|
16.6
|
151°
|
|
Influenza B
(Lee)
|
123, 185, 232,
283, 356, 416
|
409
|
0.57
|
3.7
|
-0.01
|
19.5
|
150°
|
|
Human
Parainfluenza virus-3
|
198, 260, 328,
415, 484, 536
|
530
|
0.56
|
6.1
|
-0.09
|
18.0
|
107°
|
|
Simian
Parainfluenza virus-5
|
170, 231, 295,
397, 469, 529
|
523
|
0.50
|
7.0
|
-0.16
|
19.7
|
116°
|
|
Newcastle
disease virus
|
182, 242, 306,
407, 475, 535
|
526
|
0.50
|
4.2
|
-0.31
|
20.3
|
95°
|
|
Salmonella
typhimurium
|
45, 106, 185,
236, 288, 349
|
342
|
0.69
|
9.7
|
-0.14
|
21.4
|
81°
|
|
Vibrio cholerae
|
232, 299, 555,
626, 689, 747
|
740
|
0.87
|
5.1
|
-0.22
|
17.6
|
73°
|
|
Micromonospora
viridifaciens.
|
78, 107, 138,
213, 320, 377
|
370
|
0.67
|
8.1
|
-0.27
|
17.7
|
68°
|
|
Leech
|
301, 382, 548,
602, 653, 718
|
713
|
1.09
|
4.6
|
-0.05
|
17.6
|
127°
|
|
Human
|
29, 93, 168,
225, 280, 341
|
334
|
0.77
|
3.5
|
-0.37
|
21.0
|
126°
|
*clockwise viewed towards origin
The locations of the
phenolic groups of the reported catalytic tyrosines are tabulated in
Table 2.
TABLE 2
Locations of active phenolic oxygens in the tetrahedral structures.
|
Enzyme type
|
Tyr No.
|
Coordinates (Å)
|
|
|
|
x
|
y
|
z
|
|
Influenza N2
|
406
|
8.0
|
10.2
|
17.1
|
|
Influenza N9
|
406
|
8.2
|
10.0
|
17.0
|
|
Influenza B (Lee)
|
409
|
8.6
|
8.7
|
19.4
|
|
Human Parainfluenza virus-3
|
530
|
7.5
|
10.3
|
17.3
|
|
Simian Parainfluenza virus-5
|
523
|
7.1
|
10.5
|
18.6
|
|
Newcastle disease virus
|
526
|
9.9
|
9.2
|
17.2
|
|
Salmonella typhimurium
|
342
|
4.5
|
12.9
|
20.8
|
|
Vibrio cholerae
|
740
|
5.9
|
10.3
|
16.4
|
|
Micromonospora
viridifaciens.
|
370
|
3.0
|
10.1
|
16.9
|
|
Leech
|
713
|
8.2
|
13.6
|
17.2
|
|
Human
|
334
|
7.2
|
9.1
|
18.3
|
The location is highly conserved; all examples occur
within 5 Å of the average position, approximately represented
by the enzyme from parainfluenza virus-3. According to the theory of
constraint (1) the catalytic site would have to be located precisely
where compressive stress was optimal. This would account for the high
degree of conservation. Another feature that is highly conserved is
the propeller axis. The gradients and intercepts of the equations to
the axes are listed in Table 1, showing little variation. A feature
that is not conserved across the superfamily is the rotational
orientation of the propeller about its axis. These values are also
listed in Table 1. It is estimated that they are meaningful within a
range of ± 5 degrees, based on Figure 5. There appear to be
groupings based on genetic relatedness of the source organisms. In
the case of the viruses, the orientation of the propeller is strongly
linked to the need for the tether attachment to be as close as
possible to the xy plane, but it cannot be said which phenomenon is
driving the other.
Locations of tether
attachments For
four of the viral enzymes, the tether attaches to embedded protein
chain by a disulfide bridge. These cases are listed in Table 3,
together with the coordinates of the alpha carbon of the tethered
(N-terminal) cysteine.
TABLE 3
Locations of tether attachments in the tetrahedral structures.
|
|
coordinates of
N-terminal alpha carbon of cystine pair
|
|
|
Enzyme type
|
x
|
y
|
z
|
dist from origin (Å)
|
|
Influenza N2
|
33.5
|
19.4
|
7.4
|
39.4
|
|
Influenza N9
|
34.4
|
18.3
|
8.0
|
39.8
|
|
Influenza B (Lee)
|
37.0
|
15.4
|
10.8
|
41.5
|
|
Human Parainfluenza virus-3
|
32.3
|
31.6
|
-1.7
|
45.3
|
It can be seen that all such points of attachment are
within 11 Å of the xy plane and located more than 39 Å
from the origin, as expected if the tethers are to be able to reach
the virus surface without obstruction.
Steric clashes
During docking, clashes between
side-chains of a small number of amino acids were tolerated, as
changes to side-chain orientation involve negligible energy. The
examples of the N2 and N9 enzymes are detailed in the Methods
section. A very small number of clashes between backbone atoms were
also tolerated as these could be relieved by small changes in chain
configuration.
Effect
of rotation about propeller axis on steric clashes
Of the six degrees of freedom available
during symmetrical docking, variations of most of them would have
predictable effects on steric clashes. Only one degree of freedom
seems to be variable in nature, as shown by differences between
species, and that is rotation about the propeller axis. This rotation
was chosen to illustrate the energy minimum, represented by minimum
steric clashes, corresponding to the docked structures. One viral,
one bacterial and one animal enzyme were chosen for this study. Close
to the optimally docked structure, both backbone and sidechain
clashes were recorded, but for rotations more than 30°
away from the optimum, only backbone clashes were
recorded. The results are graphed in Figure 5.
Figure
5
Effect of rotation about the
propeller axis on steric clashes in three types of neuraminidase. In
each case the upper curve shows numbers of atoms per subunit clashing
with atoms of any kind in another subunit. The lower curves show
numbers of backbone atoms clashing with other backbone atoms. The
horizontal scale is in degrees of clockwise rotation viewed towards
the center of the tetrahedron.
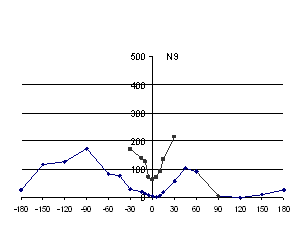
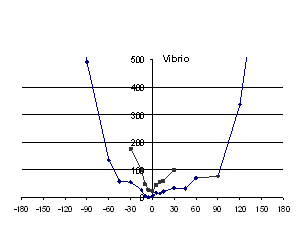
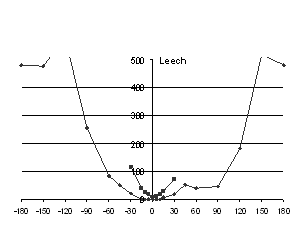
For Vibrio and Leech neuraminidases there is a
unique minimum of clashes, both backbone and sidechain, corresponding
to the optimally docked “native” structure. Note that for
Vibrio and Leech, the N-terminal domain was excluded from the
measurements as it is assumed to be incorrectly orientated in the
crystal as discussed above. For influenza type N9, there is a sharp
minimum of clashes at the “native” orientation, but also
a broad minimum 120°
away. Inspection of the latter tetramer showed
that the subunits were in an orientation where there was a large
central cavity and sparse circumferential contacts between subunits.
That is, there was very little buried surface area. While this
orientation would have low energy with respect to steric clashes, it
would have high energy because of the small buried area.
Residual internal crevices and
buried accessible surface area.
Apart from the lack of high-energy steric clashes,
a measure of the quality of fit is the size and shape of the
remaining inter-subunit crevices. According to the theory of
constraint (1), the central core of the catalytically active
tetrahedron should be tightly packed, except where space has been
left by the absence of reactants or products. The residual crevices
resulting from the imperfection of bringing the crystal structures of
the subunits together were characterized by measuring the buried
accessible surface area of the N2 and N9 tetrahedra as a function of
the radius of the probe used to measure the surface areas. To reduce
errors arising from the apparent steric overlap of side-chains, the
rotamer substitutions listed in Methods were incorporated into the
structures for these measurements. The results for the tetrahedra are
compared with the square crystal structures in Figure 6.
Figure
6. Dependence of buried surface areas on probe radius, for square and
tetrahedral tetramers of N2 and N9 neuraminidase.
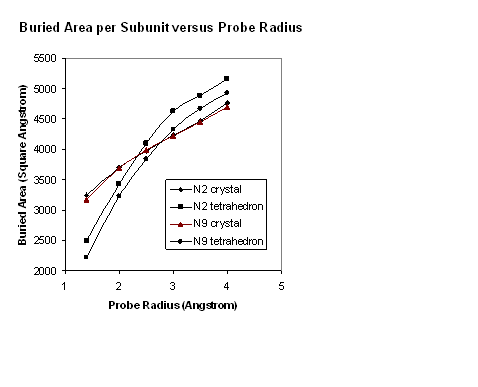
For probe radii greater than 3 Å the increase
in buried surface area with radius is similar for both the square
(crystal) and tetrahedral structures. The slope is approximately what
would be expected for a cylinder of 25 Å radius abutting a
larger structure. Because the crystal structure has no significant
internal crevices, the slope is the same at lower probe radii. It is
the residual crevices in the tetrahedra that cause the buried area to
decrease more sharply as the probe radius decreases below 3 Å.
From this it can be concluded that the residual crevices are of the
order of 2 to 3 Å across, a distance comparable to the known
uncertainty in the shape of a protein surface (3). It is assumed that
full relaxation of the tetrahedral structures would completely
eliminate the residual crevices. Such full relaxation has not been
attempted in this study, but it can be inferred from the above, that
an overall movement of each subunit of only 1-2 Å towards the
origin would allow elimination of all residual crevices. Since some
parts of the subunits are already in contact before relaxation, some
slight flexing of the whole subunit structure would have to be
included in the relaxation process.
Buried
surface area is often used as a crude measure of the hydrophobic
interaction energy between proteins. A better measure would take
account of the hydrophobic interaction that occurs even when surfaces
are not in contact (1). For the present purposes, the buried surface
areas measured by the AREAIMOL program of the CCP4 suite were
compared between the proposed native tetramers and the recorded
crystalline tetramers.
In Figure 6
it can be seen that, apart from the steep change of buried surface
area caused by residual crevices, the tetrahedral structures result
in slightly larger buried surface areas than in the crystal
structures. The conclusion that can be reached is that, even using
this crude measure of interaction energy, the proposed native
structures are as likely to exist in solution as the crystalline
structures are.
Locations of glycosylated
asparagines Many
animal proteins, and hence animal virus proteins, have
oligosaccharides attached through the amide groups of surface
asparagine residues that are part of a specific sequence (13) or
sequon (14). As a result of glycosylation experiments it is generally
accepted that all the appropriate sequons in a protein are able to be
glycosylated except in cases where the protein is synthesized without
any glycosylation, or the glycosylation system of the cell is
defective, or there is a protein structural factor preventing
glycosylation (14). Oligosaccharide side chains have never been found
inside a protein structure. Because of their hydrophilic nature, it
would seem energetically unfavourable.
For the N2
subunit, there are three widely spaced glycosylated sites
(asparagines 146, 200 and 234) (4), all of which occur on the outside
of the proposed arrangement or at the borderline between outside and
inside. Asn146 is borderline, as only low energy side-chain rotations
are needed to give the amide group clear access to the exterior. This
glycosylation site is circled in Figure 6.
For N9
there is one demonstrated glycosylated site at Asn199 (10) (listed as
Asn200 in the PDB file), which is on the outside.
For
Influenza B (Lee) there is one demonstrated glycosylation site at
Asn284 according to the PDB file, 1inf.pdb (15), and this site is on
the outside.
The
parainfluenza-3 enzyme has three glycosylated sites (16). Asn308 and
Asn351 are on the outside while Asn523 lies in the cleft where three
domains meet.
The
parainfluenza-5 enzyme also has three glycosylated sites (17), with
Asn139 and Asn267 on the outside and Asn504 borderline.
Lastly,
amongst the current collection of viral neuraminidase structures, the
Newcastle Disease Virus has four glycosylated sites (18) of which
three are included in the PDB file. Asn341 is borderline while Asn433
and Asn481 are on the outside.
In all,
there are 14 glycosylated sites in this collection of virus
neuraminidase structures and all are compatible with the tetrahedral
structures, with the possible exception of Asn523 of parainfluenza-3.
Locations of unoccupied
potential glycosylation sites
In the case of N2 neuraminidase, there
is a potential glycosylation site (Asn402) on the subunit surface
that is not glycosylated (4). With the proposed tetrahedral subunit
arrangement, Asn402 is internal and it might be supposed that this is
the reason that Asn402 is not glycosylated. However, the Newcastle
Disease Virus neuraminidase has unoccupied potential glycosylation
sites at Asn508 and Asn538 (18) and both these sites are on the
outside of the proposed tetrahedral structure. It is therefore not
possible to draw any meaning from the location of unglycosylated
sites.
Location of the hemagglutinin
site on N9 neuraminidase
Apart from its neuraminidase ability,
the N9 enzyme has the ability to agglutinate red blood cells (19) by
binding their surface polymers. If this binding is brought about by
constraint (1), such a high affinity binding site should be located
inside the tetrahedron, just as the catalytic site is. Studies of
laboratory variants (19) show that the amino acids important for
binding are 368-370, 372, 400 and 432 (N2 numbering), all of which
are located inside the proposed tetrahedral structure.
Steric and electrostatic
self-complementarity of the interfaces of influenza type N2
neuraminidase Figure
7 is a map of the three interfaces that chain A of N2 has with its
neighbouring chains.
Figure
7. Map of the subunit interface of N2 neuraminidase in the proposed
native tetrahedral arrangement. The interface consists of 3 planes
that form the sides of an irregular trigonal pyramid, the edges of
which are marked by the pseudo-threefold axes of the tetrahedron in
directions marked on the figure. The centers of the planes are the x,
y and z axes. The circled atom on the edge of the AD face is the
amide nitrogen of Asn146, a site of glycosylation. The outlined group
of residues on the AD face is the knob of large amino acids numbered
430-437. The adjacent blank area in the AD face is the depression and
cleft where catalysis is thought to occur. A disaccharide molecule in
red and grey shading is included on the pseudo-threefold axis between
the AD and AC faces where a channel appears to provide for substrate
to penetrate into the catalytic cleft. The locations of charged
groups are marked and identified.
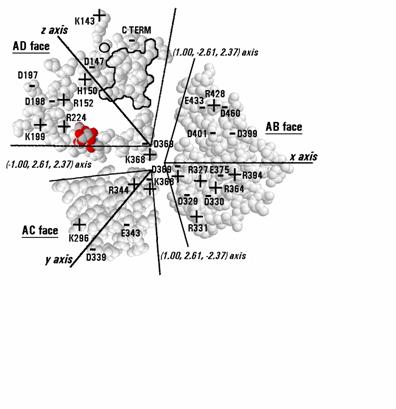
The three interfaces have been rotated into the
plane of the page. The conformation of each amino acid side-chain is
shown exactly as in the crystal structure; no rotamers have been
substituted into the structure. Each interface stretches between two
pseudo-threefold axes, and the interfaces with chains B, C and D are
centered on the x, y and z axes respectively. On this map, amino acid
residues lying along a pseudo-threefold axis appear in both of the
interfaces meeting at that axis, but are viewed from different
angles. An N-acetylglucosamine mannose disaccharide has been included
in the AD interface to show where it is thought most likely that an
oligosaccharide substrate (e.g. sialic acid - galactose -
N-acetylglucosamine - mannose - mannose (13)) would project into the
catalytic cleft. It has been fitted into a narrow channel that
connects the interior of the catalytic cleft to the surrounding
medium at the point where three subunits meet. There are, of course,
four symmetry-related channels in the tetrahedron, but only one is
shown occupied. The coordinates of the disaccharide are included in
the Supplementary
Material in PDB format. The reducing end of the mannose is
located outside the enzyme and the non-reducing end of
N-acetylglucosamine is located inside the catalytic cleft where it
would connect through a galactose residue to the terminal sialic
acid. Amino acid residues 368 and 369 lie at the center and appear in
all three interfaces.
For
self-complementarity, the half of each interface lying to the left of
its central axis should be complementary to the half lying to the
right. On the AB and AC interfaces, the steric features are no bigger
than the uncertainty in orientation of side chains, so these
interfaces provide no evidence for or against steric
self-complementarity. However, the AD interface contains the
catalytic cleft on one side of the z axis and a corresponding knob of
large amino acids (residues 430-437) on the other side, as marked on
the map. This knob partly protrudes into the cleft and would protrude
even further if the subunits were relaxed together. There are
differences in the shapes of the cleft and the knob, but these are
comparable to the uncertainty in orientation of side-chains. The
cleft and knob features are evidence of self-complementarity and
occur in similar positions in both N2 and N9 enzymes.
There are a
number of charged groups in the interfaces and these could provide
strong evidence of self-complementarity if we knew what to look for.
Buried charged groups, even when paired as a salt bridge, are
destabilizing (20, 21), although uncompensated charges are much more
destabilizing than bridged charges. On theoretical grounds, the
force-separation relationship of a salt bridge across an interface is
expected to be repulsive while water is being displaced from the
charges by close approach of the protein interfaces,
but attractive once water has been displaced. This kind of
force-separation relationship could be important for enzyme function
(1). On theoretical grounds, a close pair of salt bridges, or a
quadrupole, would be expected to be less destabilizing than two
separate salt bridges and, extending this line of reasoning, a zone
of ionic structure would be still less unstable (20). Various
arrangements of charged groups could be employed to produce a
functional force-separation relationship and these considerations do
not necessarily lead to an expectation of charge self-complementarity
across the interfaces. On the other hand, when we consider possible
lateral sliding of the interfaces, an argument for
self-complementarity becomes clear. Assuming that lateral sliding of
the interfaces would produce a non-functional structure, and that
thermal motion is continually disturbing the structure, it is
reasonable to expect that Natural Selection would have provided
surface features that oppose and correct any sliding. The steric
features described above are an example. Under sliding motion, there
is no change in the dielectric constant of the environment of charges
and therefore no doubt that opposite charges are strongly attracted.
Hendsch and Tidor (20) proposed that the role of buried salt bridges
is to produce structural specificity. Therefore it is reasonable to
expect that a native structure will exhibit a number of examples of
opposite charges in contact with each other across the interfaces.
The AB
interface of N2 contains two sets of two symmetry-related zones of
ionic structure with contributions from both subunits. It appears
that the signs and locations of the charges are such as to provide
nearly equal numbers and even distributions of positive and negative
charges. The two different zones are Glu433, Arg428 and Asp460 of
chain A with Arg331 of chain B, and Arg327, Asp329, Asp330, Arg364,
Glu375, Arg394 of chain A with Asp401 and Asp399 of chain B. Because
of uncertainty about the side-chain orientation of Asp329, it is not
clear whether this zone is contiguous with the central ionic zone
comprising the four sets of lys368 and asp369 pairs.
The AC
interface contains only two sets of pairs of opposite charges. One
pair, Asp339 and Lys296, is exposed to the solvent and therefore in a
high dielectric environment. This would reduce the force of
attraction. The second pair, Glu343 and Arg344, have their charged
groups widely separated in the crystal structure although their alpha
carbons are adjacent in the chain. Arg344 in particular seems free to
adopt many other rotameric structures and it is not clear why its
charged group is located where it is in the crystal structure. If the
subunits were brought together as proposed, this separation of
charges would become very unfavourable and it is likely that Glu343
and Arg344 would form a salt bridge and possibly a quadrupole with
their symmetry-related pair. It is worth recalling that the adjacent
Asn342 is involved in an apparent steric clash with its
symmetry-related counterpart. Therefore, bringing the subunits
together as proposed would affect the locations or orientations of
the sequence 342, 343 and 344. However, this argument is somewhat
speculative and it is conceded that this part of the crystal
structure does not provide strong evidence for electrostatic
self-complementarity.
The AD
interface contains the catalytic cleft and the 430-437 knob. The
large number of charged groups in these two features (the knob has
one negative and three positive charges) have not been included in
the map as they doubtless have substrate binding and catalytic
functions and may not conform to self-complementarity. Furthermore,
it is difficult to represent these features on a two-dimensional map.
Outside of these features, there are a number of examples of
electrostatic self-complementarity. The C-terminus carboxyl group of
Ile469 on one subunit contributes to a zone of ionic structure with
Arg224, Lys199, Asp198, and Arg152 on the other subunit. Asp197 and
Lys143 form an interfacial salt bridge in a region exposed to
solvent. Therefore they would be weakly attracted. The two
symmetry-related pairs of His150 and Asp147 form a quadrupole in the
center of the interface. This would be strongly attractive. In other
strains of influenza, other residues are present at position 147 and
it is not clear which negatively charged amino acid, if any, is
compensated by His150 in these cases. Because the pKa of histidine is
close to physiological pH, burial of an uncompensated histidine
involves loss of the charge on the histidine and very little energy
change. Therefore an uncompensated histidine at an interface would
provide no significant barrier to intersubunit contact.
A destabilized mutant of
influenza type N9 neuraminidase McKimm-Breschkin
et al. (22) and Colacino et al. (23) discussed a mutant of N9
neuraminidase in which Glu119 in the catalytic cleft was mutated to
Gly119. This mutation had the effect of relieving inhibition of the
enzyme by the sialic acid analog, 4-Guanidino-Neu5Ac2en. It also had
the effect of causing progressive dissociation of tetramers into
monomers (23) and the loss of susceptibility to binding by the
monoclonal antibody NC10 (22). As the Glu119 charged group is remote
from any of the interfaces found in the crystal structure, there was
some difficulty in explaining its destabilizing effect. In the
crystal, the nearest part of a neighbouring subunit is Trp456, 12.65
Å away, atom center to atom center, through protein structure.
McKimm-Breschkin et al. (22) suggested the destabilisation was part
of a general denaturation brought about by an entropy effect
associated with glycine residues. Colacino et al. (23) suggested that
the destabilisation resulted from mechanical transmission of a change
of conformation through the protein structure to the interface.
However, examination of the currently proposed native structure of N9
provides a more plausible explanation. As the structure is presented
here, the charged group of Glu119 is only 7.1 Å away from the
430-437 knob of the adjacent subunit across a crevice. Since a 3.6 Å
separation of atom centers constitutes contact, this means that
Glu119 is only 3.5 Å away from intersubunit contact as the
structure is presented. If the proposed structure were relaxed
together, it seems likely that each subunit could move 1 -2 Å
closer to the origin, as discussed earlier, and Glu119 could be in
contact with the 430-437 knob of the adjacent subunit. In N9, the
430-437 knob has an overall single positive charge. Therefore, a
mutation that removed the negative charge at position 119 could
easily bring about destabilisation of the tetramer. It is also
possible to propose an explanation for the fact that tetramers of the
mutant enzyme were formed in the first place. It could be that
molecules, such as oligosaccharides, bound within the catalytic cleft
affected the stability of the mutant tetramer. Storage or processing
conditions that progressively changed the nature of the bound
molecules could have resulted in progressive destabilisation of
tetramers that had been stable at the time of synthesis.
The occurrence of square
arrangements in crystals and electron micrographs of influenza
neuraminidase
Adsorption is a process common to
electron microscopy and crystal growth. In production of protein
crystals, the first step is nucleation. After that, further protein
molecules adsorb to the nascent or growing crystal, taking up ordered
orientations that allow an X-ray diffraction pattern to be recorded.
In transmission electron microscopy, the proteins are first adsorbed
onto a polymer film suspended across a copper grid. They are then
dried and stained. Because the proteins are not closely packed, the
proteins can take up orientations that are random in two dimensions
(the plane of the polymer) but tend to be ordered in the other
dimension because of interfacial forces.
When
influenza virus neuraminidase is freed from the virus with detergent
and fixed onto a surface for electron microscopy, the individual
catalytic subunits dissociate while the anchor regions at the
N-terminal of the tether remain associated, giving a characteristic
"rosette" adsorbed flat against the polymer surface (19).
However, if the enzyme is freed by enzymic cleavage of the tethers
rather than detergent, a square packed arrangement of subunits is
observed (23) and, because of the interfacial forces, all the squares
lie flat against the polymer film. The uniform occurrence of square
structures has been tentatively taken to support the idea, also
suggested by the crystal structure, that the square arrangement is
the native conformation (19). However, given that the method of
specimen preparation for electron microscopy involves an adsorption
step similar to the process of crystal growth, the occurrence of
squares is not a fully independent observation. Indeed, it is well
recognized by electron microscopists that the adsorption step,
together with the drying and staining steps in that technique,
usually generates artefacts (24). Therefore the absence of
tetrahedral forms in electron micrographs does not preclude the
possibility that tetrahedral forms occur in solution.
Comparison with the
theoretical model structure
While the discovery of these
tetrahedral arrangements confirms the applicability of constraint
theory to this enzyme family, there are differences from the model
structure (1).
Firstly,
the radius of the region of high compression (1), as shown by contact
between subunits, was 25 Å in this real enzyme whereas the
simple model predicted it only needed to be at least 6.3 Å.
Perhaps this is because the substrate is particularly large in this
case.
Secondly,
the real enzyme is far from spherical. Whereas the model involved
segments shaped like flattened quarterspheres, the real enzymes have
segments consisting of lobes protruding into the aqueous environment.
This difference is very important. It was assumed that the protein
material of the model enzyme had sufficient cohesive strength to
redirect inter-subunit hydrophobic attraction into centrally focussed
compressive stress. However, hydrophobic interactions are merely a
manifestation of the surface excess chemical potential of surrounding
water (1) and therefore the hydrophobic contribution to the strength
of a protein subunit depends inversely on its proximity to other
subunits. The structure of the real enzyme suggests that this factor
dictates a relatively large distance between segments, of the order
of 25 Å rather than the 6 Å assumed in the model (1).
However, the presence of auxiliary domains in many of the
neuraminidase enzymes would tend to reduce the average distance
between segments and it is unclear what advantage or function they
might have. They might produce subtle variations in the pattern of
compressive stress inside the enzyme.
Implications for development
of anti- influenza drugs and research into influenza antigenicity.
The X-ray structure of the catalytic
cleft of neuraminidase has been used to help in the design of
inhibitors of the enzyme as a treatment for influenza (25). The
inhibitors are analogs of sialic acid, one of the products of the
enzyme. The present work suggests that the X-ray structure of the
cleft is not complete in that it lacks the contribution of the
430-437 knob. However, the X-ray structure probably resembles quite
closely the form of the catalytic cleft that initially encounters the
substrate; that is, the cleft is open to the surrounding medium and
relatively unconstrained. It may be that the X-ray structure has been
quite helpful in designing the analogs, but a more accurate model of
the catalytic cleft in action could lead to further possible designs
of inhibitors, potentially with more efficacy as treatments.
It also
seems that research into the antigenicity of influenza has been
unnecessarily narrowed by the belief that the square arrangement of
subunits was the native form. Parts of the enzyme surface were
considered inaccessible to antibodies (19) and work with monoclonal
antibodies and escape mutants has been interpreted on the assumption
that the "top" surface of the square was accessible to
antibodies (19, 20). The interaction between an antigen and antibody
can be disrupted by a mutation within the epitope on the antigen, or
at some other site that disrupts the epitope. This suggests that, for
epitopes lying partly on one subunit and partly on another, a
mutation causing dissociation of the subunits would disrupt the
antibody- antigen interaction. This appears to be the simplest
explanation for the observations of McKimm-Breschkin et al. (22) and
Colacino et al. (23) concerning a mutant of N9 neuraminidase that
gradually lost enzyme activity and susceptibility to a monoclonal
antibody at the same time. These results are discussed in an earlier
paragraph. Although this mutant was not isolated by antibody
challenge it had the property of resistance to the N10 monoclonal
antibody, as if it was an escape mutant, and can serve as a model for
most escape mutants studied to date.
It happens
that only those antibodies that inhibit neuraminidase activity have
produced escape mutants (19, 20) and the currently proposed model of
native neuraminidase suggests that such antibodies would bind to the
exterior of the tetramer at the pseudo-threefold axis and block
access of substrate. It is therefore expected that binding of such
antibodies would be prevented by any mutation that caused subunit
dissociation, such as reported by Colacino et al. (23). Examination
of the locations of escape mutants (19) on the proposed native
interface shows that, for N2, 8 of the 9 sites are within the
interface, 7 of them within 15 Å of the origin, and 5 of those
lying along the pseudo-threefold axes. The two other sites close to
the origin and not on the pseudo-threefold axes are near the centers
of the AC and AD interfaces. For N9 (19), all of the 8 sites are
close to the origin, many also lying on the pseudo-threefold axes.
For N8 (26), 7 of the 8 sites would be close to the origin, but most
of these sites would not be close to the pseudo-threefold axes. The
eighth site lies on the bottom of the square arrangement and on the
exterior of the proposed tetrahedral arrangement and is not
considered to have a role in reducing susceptibility to the antibody
(26). It seems that all but one or two of the escape mutants must
have acted by destabilizing the tetramer and reducing the amount of
intact antigen on the virus. If this is so, the locations of these
mutations do not define the epitopes of the antibodies that selected
for them. There appear to be differences between the neuraminidase
types with respect to the regions where mutations are most likely to
cause dissociation. For N2, the susceptible region appears to be the
pseudo-threefold axes. For N9, the susceptible region is close to the
origin and close to a pseudo-threefold axis. For N8, the
pseudo-threefold axes appear to be not susceptible and the effective
mutations are located elsewhere. These sites of vulnerability could
be potential sites for action by new anti-influenza drugs, as could
the site where substrate enters the catalytic cleft.
Saito et
al. (26) measured the effect of antibody concentration on the
neuraminidase activity of their N8 escape mutant viruses. The
neuraminidase assay did not allow comparison of the relative activity
of different virus preparations. The results showed that there was
some enzyme activity remaining in the escape mutants, but not how
they compared with the parent strain. Only a study similar to that
conducted by Colacino et al. (23) could show whether the escape
mutant enzymes were largely dissociated.
Implications for X-ray
crystallography of proteins X-ray
crystallography of proteins is a series of three procedures, each
with different precision and accuracy characteristics. The aim is to
represent a native structure of a protein as a set of spatial
coordinates for each atom in the protein. The first procedure is the
production of a crystal of the protein, which will be discussed
later. The second procedure is the actual X-ray crystallography step,
in which a map of electron density in a crystal is determined by a
well-understood physical phenomenon able to be measured with high
precision and high reproducibility with suitable crystals. The third
procedure introduces further information about the sequence of the
protein and limitations on the steric arrangements deduced from other
experiments. Without the sequence, the chemical elements associated
with each center of electron density would not be known. Because this
procedure takes the form of numerical modelling, there is no limit on
the amount of precision it can generate and, because of the extra
information brought into the calculations from other measurements, it
also considerably increases the accuracy of the X-ray work.
However,
the accuracy of the crystallisation step is problematic. Each protein
crystallisation is a unique technical problem. The process of
crystallisation is a combination of complex chemical interactions,
each of them poorly understood, and it is not possible to say from
first principles to what extent any crystal structure will reflect a
structure in solution. Once a crystal has been produced and its
atomic coordinates determined with high precision, there is no other
technique that can be applied to a protein solution with enough
precision to enable the accuracy of the crystallisation to be
measured, except in rather broad terms. For larger proteins beyond
the reach of nuclear magnetic resonance methods, there have been only
very imprecise and scanty data with which to compare the crystal
structures, until now.
The work
described in this report now allows the crystal structure of
neuraminidases to be measured against themselves and the accuracy of
these particular crystallisations can be estimated. The native
conformation of neuraminidase was reconstructed by introducing
further information about biological and biophysical principles and
discarding less than 1% of the information contained in the PDB file.
The discarded information was the symmetry information, the location
of the origin and the orientations of certain surface amino acids. It
could be said that the crystallisation of neuraminidase was of the
order of 1% inaccurate in terms of transmitting information about
structure. Because each protein crystallisation is unique, it may not
be possible to extrapolate that estimate of inaccuracy to another
protein, but it seems there is only a low level of inaccuracy.
Unfortunately, the inaccurate information has affected our knowledge
of the overall shape of the proteins and locations of interfaces and
this has held back the understanding of function.
REFERENCES
(1)
Vanselow, D G. 2002. Role of constraint in
catalysis and high-affinity binding by proteins, Biophys. J. 82:
2293–2303.
(2)
Roggentin, P., R. Schauer, L. L. Hoyer, E. R. Vimr. 1993. The
sialidase superfamily and its spread by horizontal gene transfer.
Mol. Microbiol. 9: 915-921.
(3)
Del Carpio Munoz, C. A., T. Peissker, A. Yoshimori, E.
Ichiishi. 2003. Docking unbound proteins with MIAX: a novel algorithm
for protein-protein soft docking. Genome Informatics 14: 238-249.
(4)
Varghese, J N., P. M. Colman. 1991. Three-dimensional structure of
the neuraminidase of influenza virus A/Tokyo/3/67 at 2.2 Å
resolution. J. Mol. Biol. 221: 473–486.
(5)
Guex, N., M. C. Peitsch. 1997. Swiss-Model and the
Swiss-PdbViewer: An environment for comparative protein modeling.
Electrophoresis 18: 2714–2723.
http://www.expasy.org/spdbv/
(2003)
(6)
Chavaz, L. M. G., C. Tringali, P. Fusi, B. Venerando, G.
Tettamanti, R. Kato, E. Monti, S. Wakatsuki. 2005. Crystal structure
of the human cytosolic sialidase Neu2. J. Biol. Chem. 280: 469-475.
(7)
Crennell, S. J., E. F. Garman, , W. G. Laver, E. R. Vimr, G.
L. Taylor. 1993. Crystal structure of a bacterial sialidase (from
Salmonella typhimurium LT2) shows the same fold as an influenza virus
neuraminidase. Proc. Natl. Acad. Sci. USA 90: 9852-9856.
(8)
RASMOL version 2.6-ucb. 1995. Written by Roger Sayle, Glaxo Wellcome
Research and Development, Stevenage, Hertfordshire, U.K. and
enhanced by the University of California,
Berkley.
(9)
Collaborative Computational Project, Number 4. 1994. The CCP4
suite: programs for protein crystallography. Acta Cryst. D50:
760–763.
(10)
Tulip, W. R., J. N. Varghese, A. T. Baker, A. van Donkelaar,
W. G. Laver, R. G. Webster, P. M. Colman. 1991. Refined atomic
structures of N9 subtype influenza virus neuraminidase and escape
mutants. J. Mol. Biol. 221: 487–497.
(11)
Waszkowycz, B. 2002. Structure-based approaches to drug design
and virtual screening. Curr. Opin. Drug Discovery Dev. 5: 407-413.
(12)
Carlson, H. A. 2002. Protein flexibility is an important
component of structure-based drug discovery. Curr. Pharm. Des. 8:
1571-1578.
(13)
Hubbard, S. C., R. J. Ivatt. 1981. Synthesis and processing of
asparagine-linked oligosaccharides. Ann. Rev. Biochem. 50: 555–583.
(14)
Mills, K., P. B. Mills, P. T. Clayton, N.
Mian, A. W. Johnson, B. G. Winchester. 2003. The underglycosylation
of plasma a1-antitrypsin
in congenital disorders of glycosylation type 1 is not random.
Glycobiology 13: 73-85.
(15)
Sudbeck, E. A., M. J. Jedrzejas, S. Singh,
W. J. Brouillette, G. M. Air, W. G. Laver, Y. S. Babu, S. Bantia, P.
Chand, N. Chu, J. A. Montgomery, D. A. Walsh, M. Luo. 1997.
Guanidinobenzoic acid inhibitors of influenza virus neuraminidase. J.
Mol. Biol. 267: 584-594.
(16)
Lawrence, M. C., N. A. Borg, V. A. Streltsov, P. A. Pilling,
V. C. Epa1, J. N. Varghese, J. L. McKimm-Breschkin, P. M. Colman.
2004. Structure of the
haemagglutinin-neuraminidase from human parainfluenza virus type III.
J. Mol. Biol. 335: 1343-1357.
(17)
Yuan, P., T. B. Thompson, B. A. Wurzburg,
R. G. Paterson, R. A. Lamb, T. S. Jardetzky. 2005. Structural
studies of the parainfluenza virus 5 hemagglutinin-neuraminidase
tetramer in complex with its receptor, sialyllactose.
Structure 13: 803-815.
(18)
Panda, A., S. Elankumaran, S.
Krishnamurthy, Z. Huang, and S. K. Samal. 2004. Loss of N-linked
glycosylation from the hemagglutinin-neuraminidase protein alters
virulence of Newcastle disease virus. J.
Virol. 78: 4965-4975.
(19)
Colman, P. M., Neuraminidase enzyme and
antigen. In The Influenza Viruses. R. M. Krug, editor. Plenum Press,
New York. 175–218.
(20)
Hendsch, Z. S., B. Tidor. 1994. Do salt bridges stabilize
proteins? A continuum electrostatic analysis. Protein Sci. 3:
211-226.
(21)
Chong, L. T., S. E. Dempster, Z. S. Hendsch, L-P. Lee, B.
Tidor. 1998. Computation of electrostatic complements to proteins: a
case of charge stabilized binding. Protein Sci. 7: 206-210.
(22)
McKimm-Breschkin, J. L., M. McDonald, T. J. Blick, P. M.
Colman. 1996. Mutation in the influenza virus neuraminidase gene
resulting in decreased sensitivity to the neuraminidase inhibitor
4-guanidino-Neu5Ac2en leads to instability of the enzyme. Virology
225: 240–242.
(23)
Colacino, J. M., N. Y. Chirgadze, E. Garman, K. G. Murti, R.
J. Loncharich, A. J. Baxter, K. A. Staschke, W. G. Laver. 1997. A
single sequence change destabilizes the influenza virus neuraminidase
tetramer. Virology 236: 66–75.
(24)
Adrian, M., J. Dubochet, J Lepault, A. W.
McDowall. 1984. Cryo-electron microscopy of viruses. Nature 308:
32–36.
(25)
von Itzstein, M., J. C. Dyason, S. W. Oliver, H. F. White, W.
Y. Wu, G. B. Kok, M. S. Pegg. 1996. A study of the active site of
influenza virus sialidase: an approach to the rational design of
novel anti-influenza drugs. J. Med. Chem. 39: 388-391.
(26)
Saito, T., G. Taylor, W. G. Laver, Y. Kawaoka, R. G. Webster.
1994. Antigenicity of the N8 influenza A virus neuraminidase:
existence of an epitope at the subunit interface of the
neuraminidase. J. Virol. 68: 1790-1796.
(27) Varghese, J. N.,
V. C. Epa, P. M. Colman. 1995. Three-dimensional structure of the
complex of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase.
Protein Sci. 4: 1081–1087.
(28)
Zaitsev, V., M. von Itzstein, D. Groves, M.
Kiefel, T. Takimoto, A. Portner, G. Taylor. 2004. Second
sialic acid binding site in Newcastle disease virus
hemagglutinin-neuraminidase: implications for fusion.
J. Virol. 78: 3733-3741.
(29)
Crennell, S. J., E. F. Garman, C.
Philippon, A. Vasella, W. G. Laver, E. R. Vimr, G. L. Taylor. 1996.
The structures of Salmonella typhimurium LT2 neuraminidase and
its complexes with three inhibitors at high resolution.
J. Mol. Biol. 259: 264-280.
(30)
Crennell, S., E. Garman, G. Laver, E. Vimr, G. Taylor. 1994.
Crystal structure of Vibrio cholerae neuraminidase reveals dual
lectin-like domains in addition to the catalytic domain. Structure 2:
535-544.
(31)
Watson, J. N., S. Newstead, V. Dookhun, G.
Taylor, A. J. Bennet. 2004. Contribution of the active site aspartic
acid to catalysis in the bacterial neuraminidase from Micromonospora
viridifaciens. FEBS Lett. 577: 265-269.
(32)
Luo, Y., S. C. Li, M. Y. Chou, Y. T. Li,
and M. Luo. 1998. The crystal structure of an intramolecular
trans-sialidase with a NeuAca2®3Gal
specificity. Structure 6: 521-530.
Return to
http://nativeproteins.blogspot.com
APPENDIX 1:
NATURAL SELECTION AND SYMMETRY
The search
of candidate quaternary structures was narrowed by consideration of
the principle of Natural Selection and its corollary; that identical
complex biological structures are likely to function in identical
environments. In a tetrahedral arrangement, each subunit forms an
interface with each of the three other subunits, and the only
tetrahedral arrangements providing an identical environment for the
four identical subunits require each of the three faces of a subunit
to interact with the equivalent face on the neighbouring subunit.
That is, each face must be self-complementary. Thus the most probable
arrangements will have three perpendicular two-fold axes of symmetry,
or D2 symmetry.
APPENDIX 2: NATURAL SELECTION AND TETHER LOCATION
The
neuraminidases of virus particles are necessarily tethered to the
virus surface. The enzyme molecules would have some freedom of
movement to enhance encounter with substrate but a virus particle
having no protein synthesis capability cannot allow its enzymes to
diffuse away. The tether is often described as a stalk and is found
at the N-terminal end of the neuraminidase protein chain (17, 19).
The tether must be attached to the main body of the enzyme by one or
more strong interactions in addition to the main chain peptide bond
or else the main chain could begin to unravel as the enzyme tended to
move away from the virus through interactions with the environment,
such as diffusion or adhesion. Identification of this point of
attachment in crystal structures has to be partly subjective, since
the C-terminal end of the tether could have a fixed position in
crystals and appear to be part of the main body of the enzyme.
Fortunately, most viral neuraminidases appear to use a disulfide
linkage as the point of attachment of the tether. For example, the
stalk or tether of N2 and N9 influenza neuraminidase can be described
as extending from the N-terminal membrane anchor sequence, residues 7
to 35 (19), to residue 91. In the crystal, there are non-bonding
contacts between the tether and the body of the enzyme at most of the
residues from 82 to 91 but none of these is considered extensive or
permanent enough to be regarded as the point of attachment. Cys92 is
joined by a disulfide bond to a chain embedded in the body of the
enzyme and is the first amino acid, starting from the N-terminal, to
be unambiguously part of the body of the enzyme.
It is
considered most probable that Natural Selection would have provided
an attachment point such as to minimize the number of amino acids
used in chain synthesis for a given effective chain length. For a
tetrahedral structure with D2 symmetry, the optimum attachment points
would be on a plane parallel to the surface of the virus and passing
through the center of the tetramer. Only these points of attachment
would allow a minimum number of amino acids to provide a given
distance between the enzyme and the virus surface. D2 symmetry
requires that this plane contains two of the rotational symmetry
axes. A further requirement, if the number of amino acids is to be
minimized, is that the tether should be able to extend in an
approximately straight line from the enzyme to the virus surface.
This requires that the points of attachment be close to the outer
perimeter of the tetramer so that the tether is not substantially
deflected by the body of the enzyme. Of course, thermal motion would
mean that the four tethers would seldom be fully extended, but
whatever the degree of extension, the points of attachment described
above would minimize the number of amino acids required in the
tether.
APPENDIX 3:
TRANSFORMATIONS TO GENERATE CHAIN A OF A TETRAHEDRON
|
Source species
|
File name
|
First rotate
molecule with:-
|
Then add this
vector:- (Å)
|
Ref
|
|
Influenza N2
|
1NN2.pdb
|
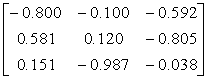
|
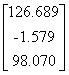
|
(4)
|
|
Influenza N9
|
7NN9.pdb
|
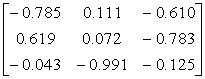
|

|
(27)
|
|
Influenza B
(Lee)
|
1INF.pdb
|
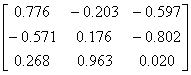
|

|
(15)
|
|
Human
Parainfluenza virus-3
|
1V3B.pdb
Chain A
|
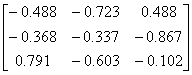
|

|
(16)
|
|
Simian
Parainfluenza virus-5
|
1Z4V.pdb
|
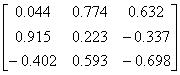
|
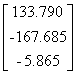
|
(17)
|
|
Newcastle disease virus
|
1USR.pdb
Chain A
|
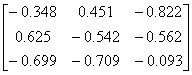
|

|
(28)
|
|
Salmonella
typhimurium
|
2SIL.pdb
|
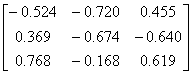
|
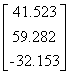
|
(29)
|
|
Vibrio
cholerae
|
1KIT.pdb
|
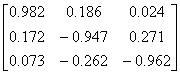
|

|
(30)
|
|
Micromonospora
viridifaciens.
|
1W8N.pdb
|
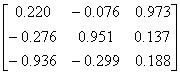
|
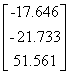
|
(31)
|
|
Leech
|
1SLL.pdb
|
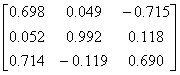
|

|
(32)
|
|
Human
|
1SNT.pdb
|
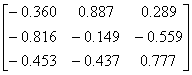
|
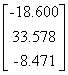
|
(6)
|
Return to
http://nativeproteins.blogspot.com